
摘要:本文介绍了除草剂L-草铵膦的一种合成工艺。在氢氧化钠的作用下,DL-草铵膦与手性辅基、NiCl2・6H2O络合形成配位化合物,该配位化合物再经水解得到L-草铵膦。反应总收率达到94%,产品光学纯度为96%。手性辅基、NiCl2・6H2O可回收利用。该制备工艺反应条件温和,操作简单,适合工业化生产。
草铵膦,又名草丁膦,化学名称为4-[羟基(甲基)膦酰基]-DL-高丙氨酸,是德国赫斯特(Hoechst)公司开发的高效、广谱、低毒的非选择性除草剂。目前市售草铵膦多为外消旋体,但只有L-草铵膦具有除草活性。国内外DL-草胺膦生产工艺已十分成熟,但L-草胺膦研究仍停留在实验室阶段,至今未实现工业化生产。因此,研究开发L-草铵膦生产工艺具有重要意义。
目前国内外报道的L-草铵膦制备方法主要有化学法和生物法。化学法以天然氨基酸为手性源或以不对称催化的方式构建手性中心,但大多存在着成本较高、光学纯度偏低等缺点。生物法主要包括生物拆分法和生物不对称合成法。生物拆分法以DL-草铵膦或其衍生物为底物,通过微生物单一降解某一构型实现拆分。生物不对称合成法以2-羰基-4-(羟基甲基膦酰基)丁酸为底物,经酶体系催化获得L-草铵膦。生物法虽然反应温和,但生物酶对生长环境要求比较严格,同时有大量的废水产生。
本研究在现有文献基础上,以(2S)-N-(2-苯甲酰基-4-氯苯基)-1-苄基-2-吡咯烷甲酰胺盐酸盐为手性辅基,在无机碱的作用下,DL-草铵膦与手性辅基、金属离子络合形成配位化合物。此过程中D-草铵膦构型反转生成L-型草铵膦,配合物经水解得到L-草铵膦,手性辅基、金属盐回收利用(图1)。
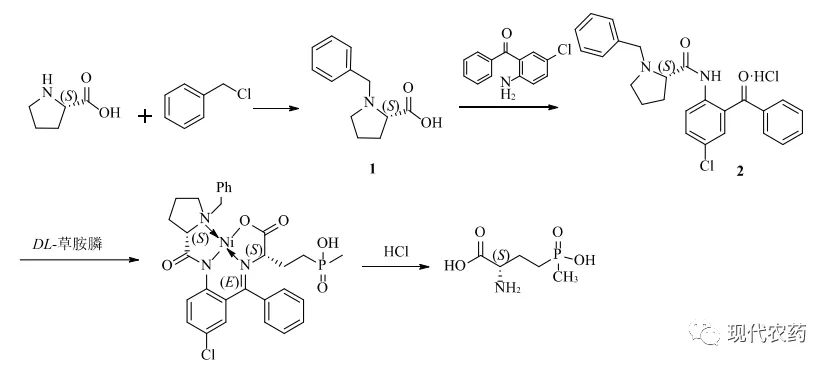
图1 L-草铵膦合成线路
1 实验部分
1.1 仪器和试剂
Bruker AV300型核磁共振谱仪(TMS为内标)、岛津LC-16高效液相色谱仪、Bruker Esquire 3000 Plus ESI/MS型质谱仪、ZF-I型三用紫外分析仪。实验所用试剂均为市售分析纯试剂。
1.2 实验步骤
1.2.1 1-N-苄基-L-脯氨酸(中间体1)的合成
500 mL三口瓶中加入甲醇200 mL、氢氧化钠19 g(217 mmol),机械搅拌下加入L-脯氨酸30.0 g(217 mmol),滴加完毕,保温反应4 h。反应完毕后降温到0℃,缓慢滴入浓盐酸至溶液pH值为3,补加甲醇150 mL,室温继续搅拌2 h。过滤除去氯化钠,滤液旋干加入100 mL乙腈打浆过滤,得到白色固体中间体1,干燥称重40.5 g,收率91.0%(以L-脯氨酸计)。
1H NMR(300 MHz,CDCl3)δ:7.44-7.31(m,5H)、4.23(dd,J=46.5、13.0 Hz,2H)、3.77(dd,J=8.6、6.7 Hz,1H)、3.62(m,1H)、2.94(dd,J=18.8、9.0 Hz,1H)、2.33-2.13(m,2H)、1.97-1.83(m,2H)。
ESI-MS(m/z):228.1[M+Na]。
1.2.2 (2S)-N-(2-苯甲酰基-4-氯苯基)-1-苄基-2-吡咯烷甲酰胺(中间体2)的合成
250 mL三口瓶中加入25 g(121 mmol)中间体1、氯苯70 mL,0℃搅拌,分批加入五氯化磷25.2 g(121 mmol),保证加入时温度不超过10℃。加入完毕,10℃继续反应1 h后分批加入28.0 g(121 mmol)5-氯-2-氨基苯酮,加入过程中温度有上升,保证不超过10℃。加入完毕后,升温至25℃反应2 h。反应完毕过滤除去固体,滤液浓缩回收溶剂及三氯氧磷,加入50 mL丙酮打浆得到25.5 g中间体2,收率95.3%(以中间体1计)。
1H NMR(500 MHz,CDCL3)δ:8.58(d,J=8.8 Hz,1H)、7.83(d,J=7.4 Hz,2H)、7.68(t,J=7.4 Hz,1H)、7.57(t,J=7.7 Hz,2H)、7.53-7.70(m,2H)、7.39-7.37(m,2H)、7.19-7.17(m,3H)、3.79(dd,J=123.6、12.9 Hz,1H)、3.36(dd,J=10.1、4.5 Hz,1H)、3.26(dt,J=8.7、4.2 Hz,1H)、2.48(m,1H)。
ESI-MS(m/z):418.1[M+H]。
1.2.3 L-草铵膦制备
250 mL三口瓶中,加入氢氧化钠6 g(100 mmol)、加入甲醇150 mL、DL-草铵膦20 g(100 mmol)、六水合氯化镍23.7 g(100 mmol),以及45.5 g(100 mmol)中间体2,缓慢升温至60℃,反应完毕后减压蒸除甲醇,加水100 mL溶解,用二氯甲烷100 mL萃取2次,合并有机相,浓缩得到红色固体。红色固体中加入3 mol/L盐酸50 mL,100℃反应3 h,红色消失,过滤得到中间体2,回收备用。滤液脱水,加入20 mL无水乙醇,静置2 h,析出L-草铵膦盐酸盐。过滤,滤液浓缩得到六水合氯化镍。L-草铵膦盐酸盐溶于甲醇,通入氨气至pH值为4~5,过滤除去氯化铵,滤液继续通入氨气至pH值为8,析出固体即为L-草铵膦,质量18.9 g,收率94.5%,ee值96.7%。
1H NMR(300 MHz,D2O)δ:3.64(t,J=5.8 Hz,1H)、1.86-1.94(m,2H)、1.34-1.58(m,2H)、1.12(d,J=13.44 Hz,3H)。
ESI-MS(m/z):182.1[M+H]。
2 结果与讨论
2.1 手性辅基(2S)-N-(2-苯甲酰基-4-氯苯基)-1-苄基-2-吡咯烷甲酰胺的合成讨论
此路线反应条件温和,后处理简单。L-脯氨酸烷基化过程中,采用氯苄代替溴苄为原料,以氢氧化钠为缚酸剂,降低了生产成本。中间体1的酰氯化反应选择五氯化磷为酰化剂,氯苯为溶剂,酰氯化产物与5-氯-2-氨基苯酮缩合后,产物中间体2直接以盐酸盐的形式从溶剂中析出,无需另加其他缚酸剂。副产物三氯氧磷可回收利用。
2.2 L-草铵膦制备的合成讨论
在原料配比、反应温度确定的情况下,考察镍盐种类对反应结果的影响(表1)。
表1 不同镍盐对反应的影响
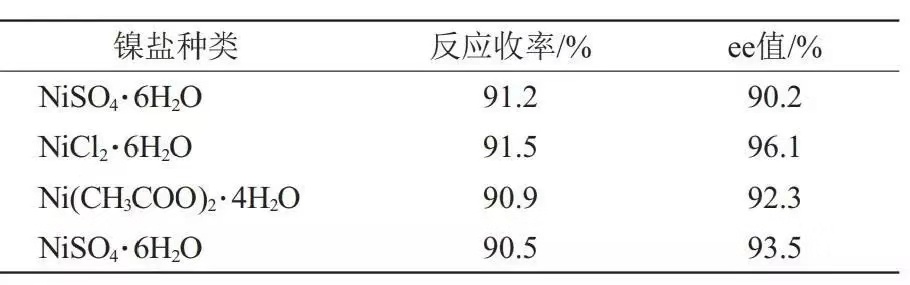
镍盐的种类对反应的收率影响不大,但对产品的光学纯度有较大影响,其中,氯化镍效果最好,光学纯度在96%以上。
2.3 L-草铵膦液相图谱
根据液相色谱图,L-草铵膦保留时间13.569 min,D-草铵膦保留时间为16.743 min(图2)。

图2 L-草铵膦液相图谱
3 结论
本研究以外消旋草铵膦为原料,在氢氧化钠的作用下与手性辅基、NiCl2・6H2O络合形成配合物。在此过程中D-草铵膦构型发生翻转生成L-型草铵膦,再经水解得L-草铵膦,回收手性辅基、NiCl2・6H2O,反应收率达到90%,光学纯度为96%。该合成路线简单,反应条件温和,适合工业化生产。
来源:王志坚, 张大永. 现代农药, 2019, 18(2): 21-23
作者单位介绍:中国药科大学 药物科学研究院


|























