
现在全球三大除草剂中,草甘膦的大面积使用已经导致抗性杂草的发生与发展;百草枯由于剧毒,导致大量中毒死亡事件发生,因而退出市场;作为仅次于草甘膦的世界第二大转基因作物耐受性除草剂草铵膦,获得巨大的发展前景。
草铵膦(glufosinate-ammonium)是一种高效、低毒、广谱触杀型有机磷类除草剂,其有效成分为posphinothricin(简称PPT),化学式为C5H15N2O4P,化学名称为2-氨基-4-[羟基(甲基)膦酰基]丁酸铵。属于谷氨酰胺合成酶抑制剂,可导致植物体内氮代谢紊乱、氨的过量积累、叶绿体解体,抑制其光合作用从而使植物死亡。
目前草铵膦的合成方法,概括起来有阿尔布佐夫合成法、高压催化合成法、低温定向合成法、盖布瑞尔-丙二酸二乙酯合成法、斯垂克-泽林斯基合成法、手性合成法等。但目前合成路线存在成本较高、反应条件苛刻、步骤复杂、产生的废料多等问题,限制了工业生产。现对草铵膦的合成工艺进行了改进,探索出一条新的合成路线。
1 合成路线
本研究在现有文献和专利基础上,综合考虑原料、实验操作及反应条件等方面因素。选用价格低廉的γ-丁内酯为起始原料,与溴素作用,选择性地发生α位单溴取代生成α-溴-γ-丁内酯,然后与氨水进行氨基化反应,加盐酸回流得到α-氨基-γ-丁内酯盐酸盐,氨基保护得到(2-氧代四氢呋喃-3-基)氨基甲酸乙酯,该中间体经氯化开环后和甲基亚磷酸二乙酯进行Arbuzov反应,经酸化水解、氨化得到产物草铵膦。结构经核磁氢谱及高分辨质谱确证。合成路线如下图所示。
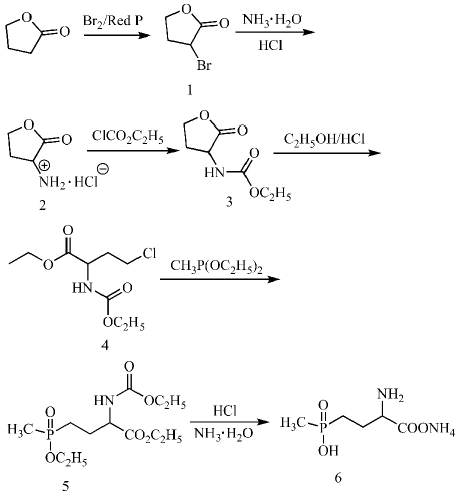
图1 合成路线
2 实验部分
2.1 主要仪器与试剂
瑞士Bruker 600 MHz核磁共振仪(CDCl3、DMSO-d6、CD3OD、D2O为溶剂,TMS为内标);RY-1熔点仪;ZF-20D暗箱式紫外分析仪。γ-丁内酯(化学纯,武汉远成共创科技有限公司):红磷(化学纯,成都博瑞特化学技术有限公司);液溴(化学纯,国药集团化学试剂有限公司);氯甲酸乙酯(化学纯,武汉格奥化学有限公司);饱和的HCl乙醇溶液为自制;甲基亚磷酸二乙酯(化学纯,湖北巨胜科技有限公司);所用试剂和溶剂均为试剂级。
2.2 实验步骤
2.2.1 α-溴-γ-丁内酯(1)的合成
冰浴下,往三口瓶中的50 g γ-丁内酯(0.58 mol)和6.7 g红磷(0.21 mol)的混合物中缓慢滴加33 mL液溴(0.61 mol),然后升温至70 ℃,继续滴加33 mL液溴(0.61 mol)。滴加完毕后,升温至80 ℃反应3 h。停止加热并冷却至室温,持续吹入空气,将过量的液溴和生成的溴化氢吹走。然后再升温至80 ℃,缓慢滴加10 mL水,等反应缓和后再加入70 mL水,回流4 h。冷却后分层,水层用二氯甲烷萃取,合并有机层,无水硫酸镁干燥。减压蒸去溶剂后得到粗产物,经减压蒸馏精制(沸程为125~127 ℃/1.7kPa),得淡黄色油状液体85 g,收率88.7%。1H NMR(CDCl3) δ:4.48(m,1H),4.41(m,2H),2.84~2.74(m,1H),2.47(m,1H)。
2.2.2 α-氨基-γ-丁内酯盐酸盐(2)的合成
在冰浴条件下,将50 g的溴代产物α-溴-γ-丁内酯(0.3 mol)缓慢滴加到270 mL的氨水(25wt%~28wt%)中,剧烈搅拌,反应48 h后,减压蒸馏除去溶剂,随后加入浓盐酸38 mL,然后在110 ℃下回流1 h,抽滤后烘干得到48.25 g的白色固体,收率69.1%。1H NMR(CH3OD)δ:7.40(s,2H),4.55(s,1H),4.46~4.37(m,2H),2.87~2.61(m,1H),2.41(dt,1H)。
2.2.3 (2-氧代四氢呋喃-3-基)氨基甲酸乙酯(3)的合成
在冰浴条件下,取α-氨基-γ-丁内酯盐酸盐10 g(0.07 mol),加入60 mL四氢呋喃和120 mL水混合,往其中加入17.6 g碳酸氢钠(0.21 mol),再将9.1 g(0.084 mol)的氯甲酸乙酯缓慢滴加进去反应0.5 h后,恢复至室温后继续反应2 h,减压蒸馏除去溶剂,调节pH值至酸性,用二氯甲烷萃取,随后减压蒸馏旋走溶剂,降温后有固体析出,抽滤得到无色油状液体6.77 g,收率为90%。1H NMR(CDCl3)δ:5.20(s,1H),4.47~4.12(m,5H),2.79(s,1H),2.27~2.15(m,1H),1.60(s,2H),0.87(s,1H)。
2.2.4 4-氯-2-乙氧羰基氨基丁酸乙酯(4)的合成
取10 g(0.057 mol)的(2-氧代四氢呋喃-3-基)氨基甲酸乙酯,加入到200 mL的耐压瓶中,随后将150 mL的饱和HCl乙醇溶液加入其中,升温至65 ℃反应12 h后,旋蒸掉其中的乙醇溶剂,加入少量的水,再用乙酸乙酯萃取,旋掉溶剂后,得到无色油状液体8.12 g,收率在60%以上。1H NMR (CDCl3) δ:5.29 (s,1H),4.47 (s,1H),4.27~4.18 (m,2H),4.13(d,2H),3.60(t,2H),2.34(s,1H),2.16(s,1H),1.32~1.23(m,6H)。
2.2.5 4-乙氧基-4-甲基膦酰基-2-乙氧羰基氨基丁酸乙酯(5)的合成
称取10.0 g(0.042 mol)的4-氯-2-乙氧羰基氨基丁酸乙酯与11.5 g(0.084 mol)的甲基亚磷酸二乙酯加入到150 mL无水甲苯中混合后,在氮气保护下,回流反应12 h。减压蒸馏,回收甲苯和甲基亚磷酸二乙酯,加入适量乙酸乙酯后,用饱和的氯化钠溶液洗涤3次,用无水Na2SO4干燥,减压蒸馏去除溶剂,得到9.6 g油状液体,收率为75.8%。1H NMR(DMSO-d6) δ:7.49(s,1H),4.17~3.97(m,6H),2.14~1.91(m,2H),1.65~1.07(m,13H)。
2.2.6 草铵膦铵盐(6)的合成
称取10.0 g(0.032 mol)4-乙氧基-4-甲基膦酰基-2-乙氧羰基氨基丁酸乙酯加入到100 mL 6 mol/L HCl溶液中,回流12 h。再用氨水调pH值9,继续回流8 h,减压蒸馏后,用甲醇重结晶,得到白色草铵膦晶体5.2 g,收率为80.9%。1H NMR (D2O) δ:4.00 (t,1H),2.20~2.06(m,2H),1.89~1.67(m,2H),1.39(d,3H)。
3 结论
本研究对目前已报道的多条草铵膦合成路线进行了考察,将研究重点放到了α-氨基-γ-丁内酯盐酸盐的合成上,采用了低廉的溴素作为溴源,经改良的减压蒸馏方法得到α-溴-γ-丁内酯,然后经氨基化反应,酸化得到关键中间体α-氨基-γ-丁内酯盐酸盐。本工艺大大降低了成本,反应条件温和,适合工业化生产。
(来源:农药)


|























