
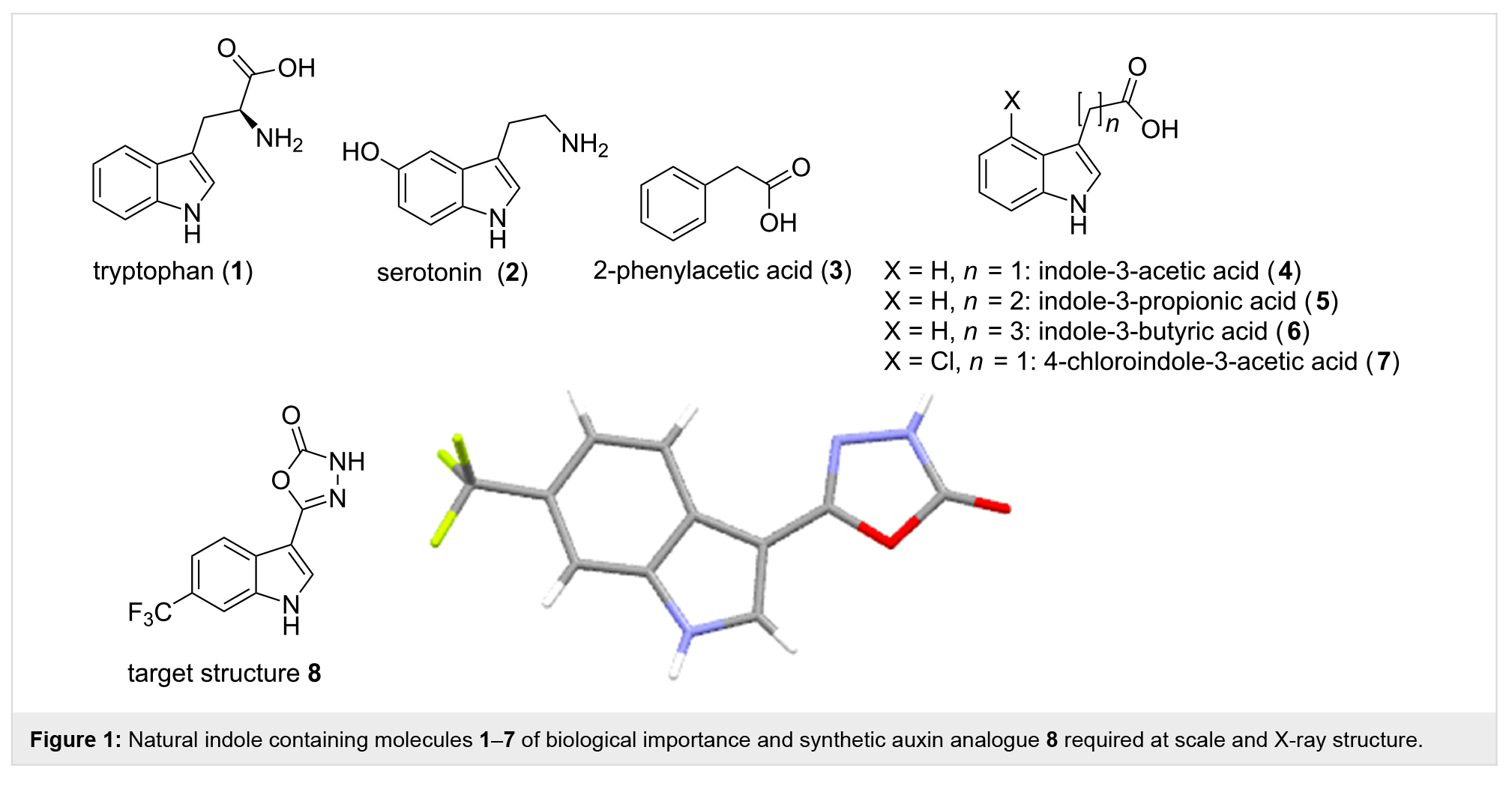
�����������ɫ����(1)����ص�����Ѫ����(2)�Լ����ิ�ӵ��������Ȼ���ʺ�ҩ���г���������Ҫ����������ӻ��ṹ֮һ��80%��ֲ�������ϳɵ��������غ�������ṹ��ͼ1���ṹ3��7����������ֲ�D���ڴٽ��͵���ֲ��������������Ҳ��������Ҫ���á��������ǵ��ڻ����Щ�ṹ�ѳ�Ϊ�о��Դٽ����������Լ�����Ŀ��ֲ�������ΪĿ������ũ�ó��ݼ���ѡ��
�����Ӣ�����״�ѧΪ�о��ϳɵ�����-3-��������������Ҷֲ�˫��Ҷֲ��е����ռ���ֲ�����Ҫ�����Ʊ��ṹ8�Խ��д��ģ��������顣��������ģʽ�ͻ�������������о������ʱ��������������ʵ������кܴ�Ӱ�죬��ˣ��о���Ա��Ϊʹ�ý�Ϊ����������ѧ�������������Ƿdz���Ҫ�ġ�
��1835���ϣ���״κϳ������������������ʮ���ֶ��ص���������ﱻ������������������Щ�м�ֵ�Ľṹ�п����»��������Ҫ�ԡ�Ȼ������������������ʺϳɵĹ�ͬ�ص�����Ҫʹ���к����ʣ����£�Fischer�����ص�����Japp-Klingemann����������Hemetsberger-Knittel�������������ỷ�γ�֮ǰ�����ڶಽ��Ӧ�й����ض����ܵ�ǰ�塣Ϊ�˿˷���ЩDZ�ڵ�ȱ�㣬Marcus Baumann�����Ŷӿ�ʼ����һ�����ԵĹ��գ��������۵ĵ���������Լ�����һ�����������������ķ�ʽѸ�ٺϳ���������ᡣ
1 Ian R. Baxendale�Ŷӹ��ںϳ����ᵥԪ���Ż�
Ϊ��ͨ���Ƚ��ĺϳɲ������ɺ������ᵥԪ�� Durham ��ѧ��ѧϵ��Ian R. Baxendale�����ŶӾ����о�������������(10)Ϊ���Լ����ڼ�鵼��SNAr��Ӧ�д���2-��������(9)��Scheme1�������õ��ļӺ���11���ڷǾ����⻯�����£�ͨ����ԭ�������������������12������Ӧ�������ܻ�����12��Ԥ���������ϻ�õ���Ӧ������13�����ͨ�������ʻ������������CDI��1,1-�ʻ���������������˫�����ȼ���̼�������������õ�����IJ�Ʒ8������1����
���ֺϳɲ��Ծ��ж������ƣ���Ϊ���������ֳɵĵ�����Լ������������������base��HCl��H2O�������ڹؼ��Ļ�����������ʹ�ù�ҵ�Ͻ��Ѻõļ���գ���سɹ�ϲ�ˣ�Beilstein Journal of Organic Chemistry 2017, (13), 2549-2560����

1.1 ��Ӧ�ܼ���ɸѡ
Ϊ�˿�ʼ�����о������Ƚ�����һ��ȫ���ɸѡ���̣���ȷ���γɻ�����11��������������������1����
��1 ���ܼ�����¶Ⱥ��Լ���ѧ�����ȵ��Ż�ѡ��
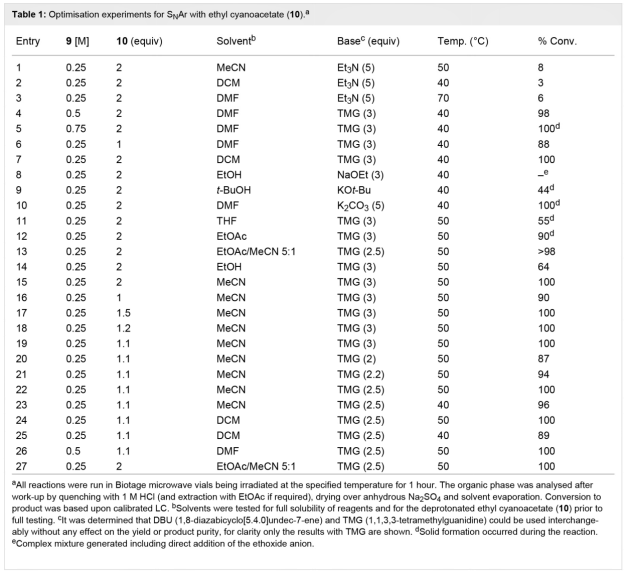
��ȻDMF�������Ǻܺõķ�Ӧ�ܼ�����1����3��6��10��15��23��26������ڴ���Ӧ��1 mol/L HCl������Ч����ȡ��Ʒ�����������ѡ����������������죬��ʹ��ȡ�dz����ף����ڷ�Ӧ�����й۲쵽��������ɫ��ճ���ij������1����12����������Գ߶ȵ�������Ӧ����������������ģ���Ϊ������ɶ�����Ȼ�����о����֣�����10%��20% v/v��������Ա�֤��Һ��ȫ���ȣ�Ҳ���Խ��м�ˮ��ȡ�������ʽϸߣ���90%����Ȼ�����ӳ������������DCM������Ч���ܼ�����1����7��24��25�������0.25 mol/L�ĵ���9��Һ��50��ʱ��TMG��DBU��2.5��������������������10��1.1���������ж���ת����
1.2 ����װ�õ��Ż�
������Щɸѡ��������һ��������װ�ã�����Ԥ����ԭ��Һ��һ����T�ͻ�����л�ϣ����ֱ�ӵ�һ��ά����50��ļ��ȵ�������Ȧ��Ӧ���У�Scheme 2��������35��108 min��ʹ��ר�û����оƬ��ϵĵ�������������1 mol/L���������γɵ����ɫ��Һ��SNAr�ӳ���������ӣ�����Ȼ�ϲ��Ļ��������롣
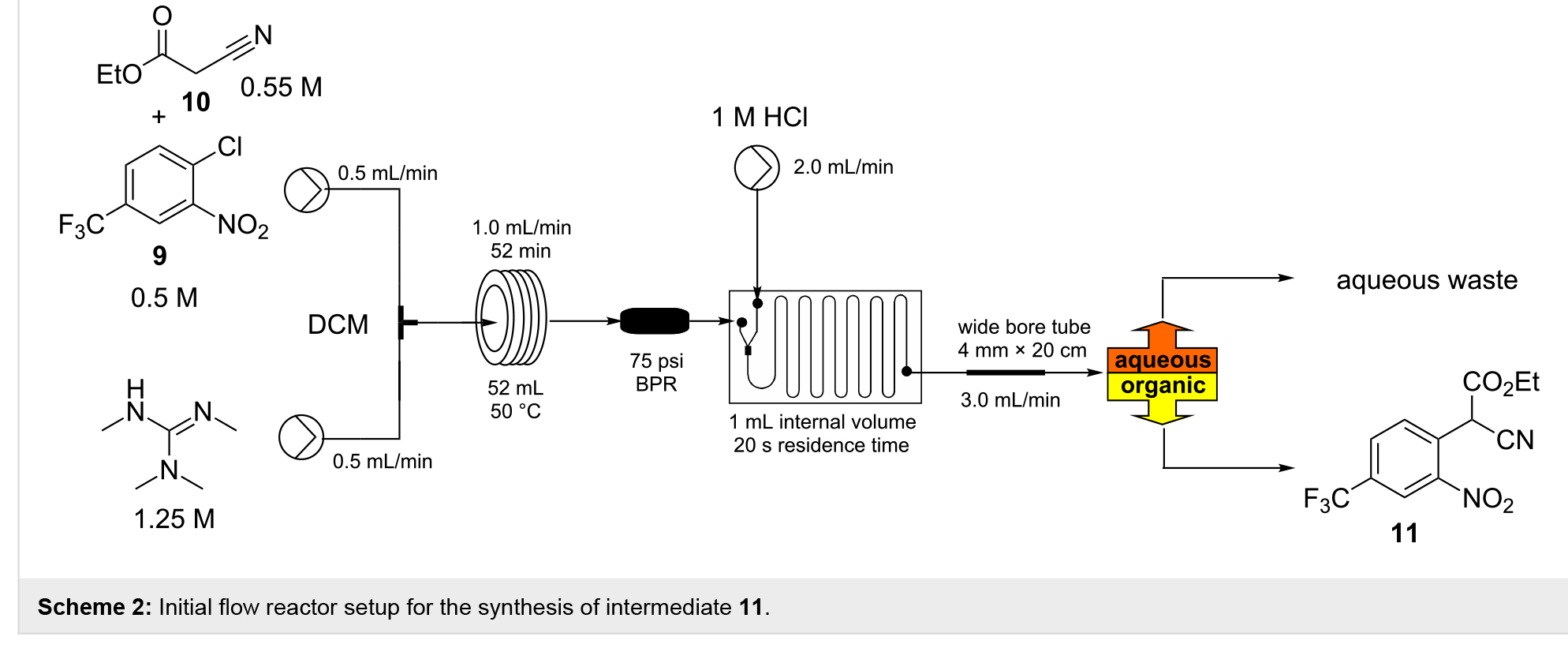
Ϊ��������룬ʹ��һ�����ڸĽ���Biotageͨ�÷������ı���Ĥϵͳ����ʹ�ý��ص��Ȼ�������Դӽϵ͵����Ӵ�ȥ���������ˮ����Դ�����λ����������������Ӧ������������Ϊ0.5��1.2 mL/minʱ����װ�ÿɿ��ؽ��������õĴ���ͷ��롣Ȼ�����ڸ������²�������������IJ���ȫ���루��Щ��Һ�γɣ������²�Ʒ�е�������ˮ����ʧ�Ӷ������ʧ�����������ڽϸߵ��������߽���оƬ�����˽ϴ�ļ��������Լ��γɸ߶�����������Ҫ���ȶ�ʱ�����������������ͨ����ˮ�ӵ�һ���������������ڶ�����Чװ�ã�����ͨ������ƽ�з�������ԭ���Ĵ������ɷֿ��������
��Ȼ��Щ��������ƻ�ʹ�÷��������ʱ�������ŵģ����ڹ������ǿ��еġ���ˣ����о����ø������õ�T�ͺ�Y���������滻������Ļ��оƬ�����������������������⣬��Ϊ����ȫ�������˲�Ʒ���ղ�������ص���Ⱦ��һ�ָ�ֱ�ӵķ������ڷ�����֮ǰ����һ�������ֲ���������ͨ����������һ���ھ������Ĺܣ����ھ�1.6 mm��4.0 mm��20 cm�ķ�Ӧ����չ��������ʵ�ֵġ���ʹ�÷�Ӧ������ͨ��2.0 mL������������1.0 mL���ϵ�����������ϵͳ�����ӵڶ���52 mL�������̹ܣ��ɹ��ؽ��д���Ӧ��Ȼ���ɵ���Biotageͨ�÷��������δ���������������£�����Ӧ���ij��϶���ʾ������ת����ֻ�����ܼ��������Ʒ�������������Ӧ����������Ϊ1.8 mL/min����ʹ������̬�����µ�����������ﵽ27 mmol/h��
���ܸ÷�Ӧ�����ܶ�������������Ч�ʵ�������Ҫ���ҵ��ǣ�DCM��֤���������⻯������һ�������ݵ��ܼ�����Ȼ�ܼ������ǿ��ܵģ�������ȷ�����ǣ���������������Ϊ�ܼ�����������Ϊ���Ӽ����������Ҵ�ϡ��ʱ����ʹ����Ļ�ԭ���������Գɹ����С��ڴ˻����ϣ��о��������������м���11�ķŴ�Ӧ����֮ǰ�ɹ�ʹ��Coflore��AM������˾ACR�豸���������ཬ�����߾����ںϳ���ʹ�ø��豸���˷����ܽ��Է������������⡣
1.3 Ŀ������ṹ8�ĺϳ�
��ʼ���������������зֱ�����Һ���Ʊ��������ϣ������Ǽ������������(10)�ڼ��뷼����(9)����Һǰ��ϣ�ע������һ�����ȹ��̣���Ȼ�����ACR����Ƶ������Ϊ8 Hz��Scheme 3����

Ϊ�˼�ǿ��һ���̣����Լ����ܼ���Ϊ����Ŀ�꣬�Ա������Ļ�ԭ���裨�Ҵ�ϡ�ͺ��б���һ�����еķ�ӦŨ�ȣ����߽�����һϵ��Ũ�Ⱥ������Ż������շ��֣���3 mL�Ļ�������£��൱��ͣ��ʱ��ԼΪ30 min����Ӧ���¶�Ϊ65�棬�ܹ�ʵ��1.5 mol/L����9��Һ�Ķ���ת������������ʱ����Ӧ���ij���Ϊ����ɫ����Һ�����а����������ԼΪ10%�Ĺ��壬����ʹ�ڽϳ�ʱ��������14 h��Ҳ�ܺ�����ͨ��ϵͳ���д�������װ�õ����۹���Ч��Ϊ0.108 mol/h����������Ϊ�˱����������ͺ���������6 mL/min���������͵�2 mol/L HClˮ��Һ�Ļ�ϵ�������һ����̬���Ԫ����Scheme 4����ͨ��������ɫ�ķ�Ӧ�����ת��Ϊ�ռ�ʱ����������dz��ɫ˫����Һ�����������۲쵽��Ч����ļ���ͨ�����л����1H NMR��LC������ȷ���˸÷�������ɹ�����ʾֻ�в���11����������������10�����������˹����룬Ȼ����Na2SO4�ϸ�����ܼ����������õ���Ŀ�����11������94%��96%������ÿ��20 minȡһ��������ȡ6�Ρ�

����ʵ�ּ�Ъ�����Զ�����ʵ���ҷ����Ѿ��б��������û����Ӿ�ϵͳ�Ͳ��ù�ѧ���Ӧ�絼�ʣ��迹��������ȷ����λ���ֵ���������豸��ȷ������롣Ȼ���������ض���Ŀ��ʱ�����Ʊ���ʹ�ø��˹��ķ��������϶��Ὣ���ֽ�ʡ�Ͷ������豸����δ���ķ�չ�ƻ�������ϵͳ��ʹ��һ���ļ�Ъʽ�ռ�����������λ�ú��ʵĸ�ЧҺ��ɫ�ױ���������ȥ��ˮ����л��ࡣż�����˹���Ԥ������Ъ����У�ã��Ա��ֹ��������������һ�����ʵ���ν���ȥ����ͬ�������ܼ�������Ŀ�����11�ķ������ʸߴ�93%��97%��
��2
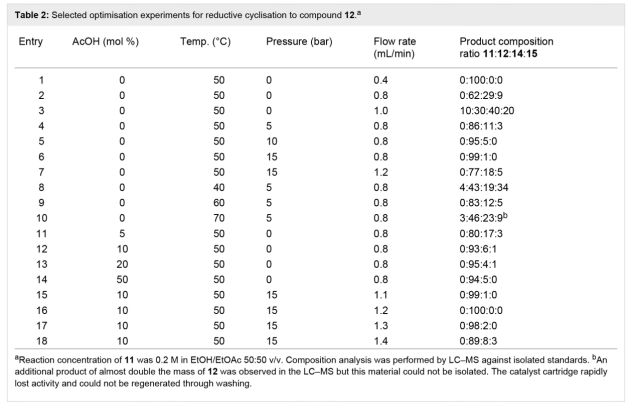
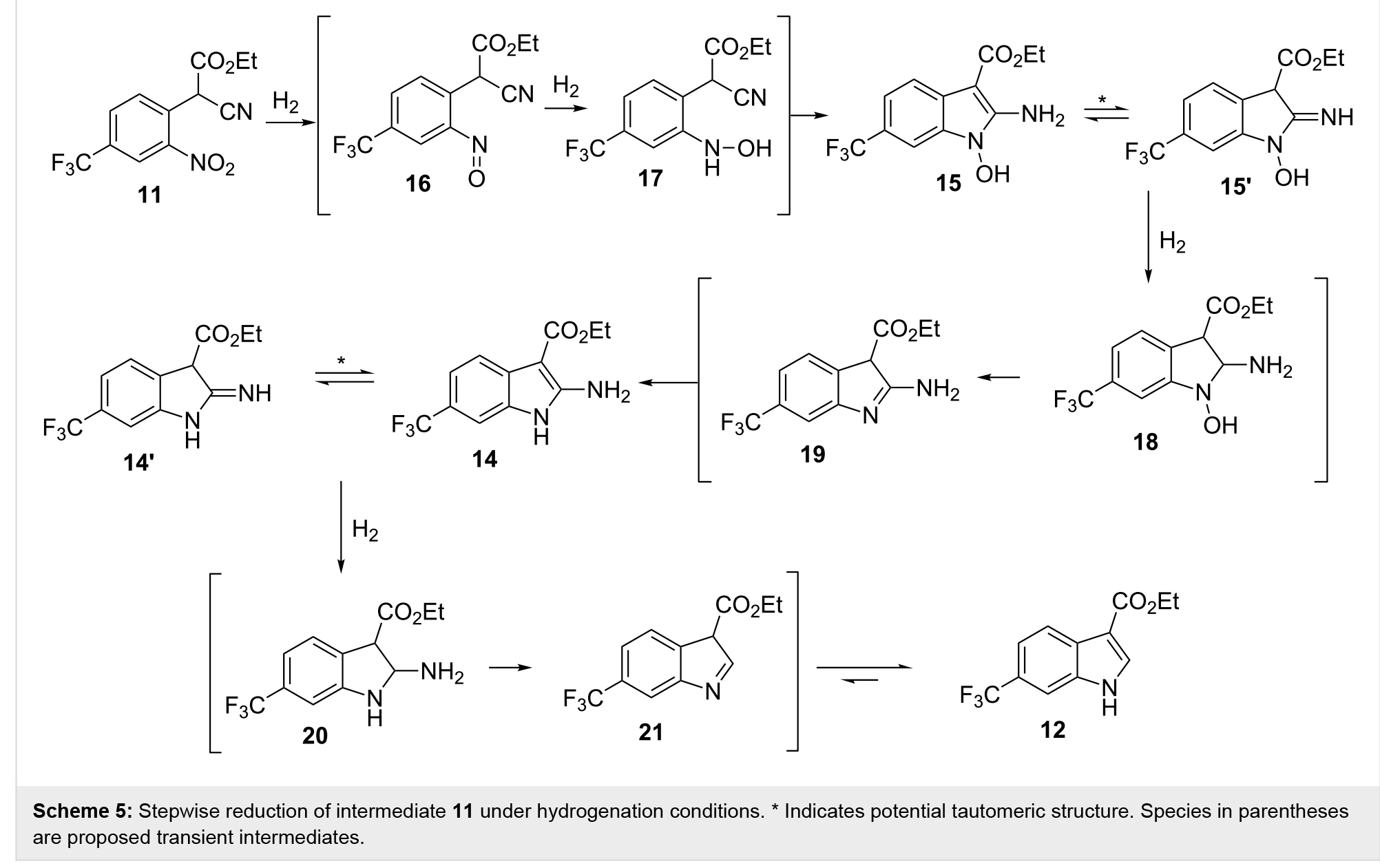
ˮ����������ռ�����Һ�к���0.4 mol/L��Ʒ11���ò�Ʒ11�����Ҵ�ϡ�ͣ����ṩ0.2 mol/L��ԭҺ��������һ����ԭ�Գɻ����衣��ȫ�������£�ʹ�ú�10 mol% Pd/C������ThalesNano H-cubeϵͳ���л�ԭ������Ϊ0.4 mL/min���¶�Ϊ50�棬��ȫת��Ϊ�������12����2����1�����������ߵ�0.8��1 mL/minʱ���۲쵽ԭ����ȫ���ģ����������ϲ����2����2��3���������ɵ���ϸ����������������12��62%�����������ֻ����ͼ2���Ĵ��ڣ������ֻ������������������ֱ�ȷ��Ϊ�������ᣨ14��29%����n-�ǻ��������ᣨ15��9%��������Ԥ�ڣ��������ٽ�һ�����ӣ�n-�ǻ���������15�ı������ӣ������ֲ���ȫ��ԭ�������ࡣ��������������ʹ�ô��⻯��ѧ����������п�������鵼��ԭ���о�������������������ԭ������һ�£�Scheme 5������ʵ�������п������Կ�������Ҫ�����������������ȫ��ԭ����˽ϸߵ���Ũ�ȣ�ѹ����������ģ���2����4��10������ݻ����������������ӻ����Դٽ���ԭ���裬��������ˮ��16��18���Ͱ���20����ȥ�������ɸѡ��һϵ�����Դ���������������10mol%��30 mol%�����ѣ���2����11��14�����ǿ�����ϸߵ��Ḻ�������ᵼ�²����ϸߵ��ڲ�ѹ������ijЩ����£����۲쵽��������γɣ�����Ҫ��������ǰ��ֹ����ȫ�رա�ʹ�����Ტ�������Ũ�ȣ�10 mol%�����ڽϸߵ��ڲ�ѹ����15 bar�������У��ܹ��ȶ��������ز���������������ߵ�1.3 mL/min��ͬʱȷ������98%��ת�������������12����2����15��18�����ת��Ϊ15.6 mmol/h��3.7 g/h��Ʒ����ͨ����ͨ��9:1������/�����ܼ���������ĥ���ɷ������ɫ�������ղ������93%����ͳ�ȥ�˲��������ᡣ

ͼ2
�ڽ�������ʵ�������У��о���������̵�����������裺������ȡ�����������Ȼ��ɻ��γ�3H-[1,3,4] �f����-2-1��Ԫ��
�ڴ˹����У���������12��0.95 mol/L THF��Һ������Һ��THF�У�1.0 mol/L����ϣ�ֱ�ӽ�����ȵ�������Ȧ�У���100���½��й��ȣ�Scheme 6����ͣ��ʱ��Ϊ40 min��������ȫת��Ϊ��Ӧ������13��������13�뺬��CDI��THF�У�1.1 mol/L������һ����Һֱ�ӻ�ϣ����ڵڶ�������Ȧ��75�����26 min�����ṩ�����ղ�Ʒ8�Ķ���ת������Ʒ��ͨ��QP-SA����У�һ�ֻ��Ṧ�ܻ��ۺ�������ᴿ���ò�Ʒ8���ܼ������õ���ɫ���壨94%��������Ҫ��DCM�ؽᾧ�õ���ɫ�Ǿ���ĩ�����ȸߣ�����82%��
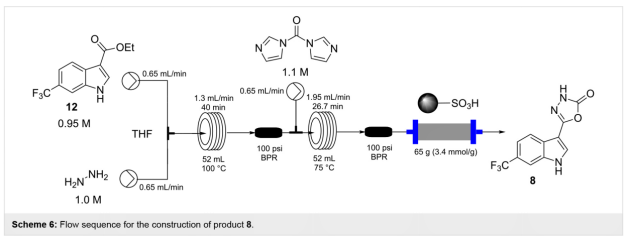
�����һ����ʹ��̼���������ΪCDI����Լ�������һϵ�еij��ԣ���һϵ�������¶�ʧ���ˣ�����ͬǰ��������γɲ�����ֱ��ʹ�ã������ʻ����£�CAS��6294-89-9�������飬Ҳʧ���ˡ�Ȼ��������������0.4���������Գɹ���Ӧ���ں��ߵijɻ����̡�ʹ����Scheme 6���Ƶķ�Ӧ������������������ȷ��У�0.8 mol/L�����м���13����Һ��ϣ�ͨ��һ�����ȣ�55�棩��������Ȧ��Ȼ��ͨ��һ������QP-DMA��N,N-�����а��۱���ϩ������䴲ɨ��Ͳ���ܼ�������ƷΪ��ɫ���壬��������91%����ʵ���У���һ������֤��ԶԶ������ǰʹ�õ�CDI����ֱ�Ӵӷ�Ӧ����ø��ߵ������ʺ����������IJ�Ʒ��ʵ���ϣ����ղ�Ʒ8�Ĵ����㹻ֱ��Ӧ�ã��Ӷ���Ӧװ���в���2 h��һ���ȶ����м���12������ģ������
2 �ܽ�
��1���������������IJ��裬�����Ѿ��ܹ�ͨ��һ������ʵ�����̿����Ʊ������Ŀ�����8��
��2��������ʵ�������У�Ӣ��AM Technology��˾�������༶���跴Ӧ��Coflore ACR�ڵ�1�������м���11�ķ�Ӧ�����˹ؼ����ã���Ϊ��Ӧ�л�����ɴ������ܵĹ����θ������Coflore ACR�����ڷ�ֹ������ͬʱʹ��Ӧ�����ȶ����У�
��3��Coflore ACR��Ӧ�����Խ����������Ӧ�еĶ������⣬�����Է�Ӧ�ĸ�Ч�����Լ������������Թ���ķ�Ӧ��������Coflore ACR��Ӧ�����õ�ʵ�����ܣ�
��4����������Coflore ACR��Ӧ��ʵ���˵�1���м���11���������ȶ�����������������������ʵ�ִ��������������Թ��岻�����������������������⣬����������ھ�ϸ������Ʒ�Ĵ����Ʊ����ؼ������ã�
��5��Coflore ACR���Ѿ�������ҵ���ķ�Ӧ�����߱��ܺõĹ淶�Ժ�ͨ���ԣ��纬�̵ȶ��ิ����ϵ�Ĺ�ҵ��Ӧ��ǰ��ʮ�ֹ�����
��Դ��������һ���Ƽ�����˾


|























