

随着欧盟农药管理新规(EC)No 1107/2009条例(代替原91/414/EEC法令)的诞生,欧盟“相同原药”认定及资料减免政策也随之发生了变化,其中包括于2011年6月17日生效的“相同原药”认定新法规SANCO/10597/ 2003-rev.9。本文对欧盟现行的“相同原药”认定及资料减免政策进行了详细介绍,为相关部门及企业提供信息参考。
因为管理体制的不同,欧盟现行的资料减免和“相同原药”认定政策与我国的相同产品农药登记制度存在一定的差异。为减少试验,特别是脊椎动物试验,欧盟执行鼓励甚至强制资料共享的政策,因此探讨欧盟“相同产品”认定政策需从其资料保护及共享政策说起。
1 欧盟农药登记资料公开及共享政策
按照(EC)No 1107/2009条例Article 57(l)的要求,欧盟各成员国需要对公众公开已登记农药制剂产品中有效成分、安全剂和增效剂的名称及含量信息。为了避免重复试验,制剂产品申请者在进行相关实验之前需确认是否有含有相同有效成分、安全剂和增效剂的产品登记,及其所用助剂(adjuvant)是否获得登记。农药管理部门可以应申请者的要求向其提供已经登记产品的资料清单,并向其提供资料拥有者的联系方式,以帮助促成资料共享,达到减少登记试验的目的。
按照欧盟有关农药登记资料豁免的规定:如果申请人提供证据(授权信或者资料已过资料保护期的证明)表明其可以使用某些已经存在的试验资料,在提交了下述三方面信息后可以免于提交相关试验报告:农药产品定性方法、100%产品组成和1份没有使用禁用助剂的声明;产品中有效成分、安全剂和增效剂的信息(如有必要,需要进行原药的相同产品认定);应评审国农药管理部门的要求,提交相关数据以比较申请产品与被引用资料产品对环境和人类健康造成的影响。
对于脊椎动物试验资料共享,欧盟有着更为严格的要求。任何农药登记申请人在进行脊椎动物实验之前,都需要提供证明,显示该项试验没有被重复进行,并要求新产品的申请者在符合条件的情况下尽量和资料的拥有者达成资料共享的一致意见,如未能达成一致,则申请者需要将相关信息告知农药管理部门,后者将通过法律途径使得二者强制达成一致意见,已达到保护动物,减少实验的目的。
2 欧盟农药登记资料保护政策
在欧盟,登记资料满足下列2个条件即可以受到保护:(l)在产品登记或对现有登记进行修改以允许在其他作物上使用时必须要提供的资料;(2)按照良好实验室规范(GLP)或良好实验规范(GEP)完成的资料。但脊椎动物实验需要在一定条件下强制共享。在申请产品登记时,申请人就必须提交需要受到保护的资料清单,否则其登记资料将不会受到保护。资料保护时间从登记之日起为10年,对满足要求的低风险农药(定义见(EC)No1107/2009 Article 47),其资料保护时间可以延长到13年。同时,为鼓励在小作物上的农药登记,对于登记后5年之内扩作到小作物上使用的,针对每一种小作物,资料保护期可以延长3个月,但总的资料保护时间不能超过13年,低风险农药不能超过15年。对于为进行重新评估或续展登记提供的资料,其保护期为30个月。资料在保护期内的,可以通过资料拥有者授权的方式使用,如果登记产品已过期,则登记资料不再受到保护。
3 欧盟农药制剂产品的平行贸易(parallel trade)政策
为了鼓励欧盟成员国之间的农药贸易,对于在欧盟成员国之间的农药进出口,欧盟制定了平行贸易政策,即如果某农药产品已在某一成员国获得登记,另一成员国有其“相同产品”登记(组分完全相同),则后者可能给该产品发放平行贸易许可证,即该产品无需在进口国再次申请登记。在同时满足下列三种情况时,产品可认为完全相同:(1)同一公司或授权的其他公司生产,工艺路线相同;(2)有效成分、安全剂和增效剂标准和含量相同,剂型相同;(3)使用相同或等同性的助剂和包装,在对环境和人类健康的影响上也具有等同性。
4 欧盟“相同原药”认定政策
4.1 “相同原药”定义 如前所述,欧盟并无“相同原药”登记类型,按照欧盟的法律规定,在下列几种情况下,需要进行原药的等同性确认(即我国的相同原药认定):(1)在制剂产品获得登记后,改变原药来源,或原药登记者增加或更换供货商(欧盟登记证持有者并非只限于生产企业,因此登记证持有者可以有多个供货来源(source));(2)改变原药生产工艺或生产地点;(3)中试生产到正式生产。因此欧盟将“相同原药”定义为:新的原药来源按照相关规定与参照原药进行比较后,新原药中的杂质有相同或更少的不良影响。
4.2 资料要求 根据上述三种情况,在进行原药的等同性确认时,需要提交的资料也各不相同。
4.2.1 原药来自一个新的或不同的生产企业 需要提供91/414/EEC附件2A中要求的1.1―1.11部分资料:1.1申请者信息,包括姓名、地址等;1.2生产商信息,包括名称、地址等;1.3 ISO通用名或建议的通用名称;1.4化学名称(IUPAC或CA命名);1.5产品开发代码;1.6 CAS、EEC和CIPAC编码;1.7分子式、结构式和分子量等;1.8有效成分的合成路线;1.9有效成分含量(用g/kg表示);1.10异构体鉴别、杂质和添加剂(例如稳定剂)及其分子结构式和含量(用g/kg表示),产品中含量超过1g/kg的成分都必须提供如下信息:IUPAC或CA命名的化学名称、ISO通用名、CAS、EEC和CIPAC编码、分子式和结构式、分子量及含量;1.11批次分析数据(总含量不得低于98%),如果有效成分在几个不同的生产点生产,则需为每个生产点生产的产品分别提供批次分析报告,必要的话,还必须同时提供中试生产的样品和正式生产样品的批次分析,和4.1部分资料有效成分、相关杂质、含量超过0.1%的杂质的分析方法,包括方法线性关系、精密度、准确度等。
4.2.2 中试生产到正式生产 需要提供91/414/EEC附件2A中要求的1.11部分资料。对于1.1―1.10部分资料,如果没有变化,申请人仅需提供一份声明。如果杂质资料发生变化或使用了新的分析方法,则还需要提供4.1部分资料。
4.2.3 生产工艺、生产地点发生变化或增加生产地点 需要提供91/414/EEC附件2A中要求的1.1―1.11部分资料,如果杂质资料发生变化或使用了新的分析方法,则还需要提供4.1部分资料。
如果在91/414/EEC附件1中有超过1个可供比对的产品标准,则需要将新的原药标准和所有已列入附录1的产品标准进行比对,且只要和其中之一认定为相同产品即可。对于参比标准的建立,欧盟SANCO/6075/2009条例中有详细规定。
4.3 认定程序及判定依据 与FAO/WHO农药相同产品比对程序类似,欧盟相同产品认定程序也分为以下2步:阶段1:对91/414/EEC附件2A中要求的1.1―1.11部分(产品化学资料)进行审核,以对产品的等同性进行评价。阶段2:如果依据阶段1提供的数据不能对产品的等同性进行评价,则需要进行阶段2的评价,即对产品的毒性/生态毒性进行评价。在阶段1中,如果满足下列标准,则可认定为相同产品:原药含量不低于参照产品;没有新的杂质;相关杂质的限量没有增加;所有非相关杂质的限量不超过下列标准(表1):
表1 非相关杂质的限量标准
|
在参照产品中非相关杂质的限量 |
可接受的增加量 |
|
≤ 6g /kg |
3g /kg |
|
> 6g /kg |
限量的 50% |
相同产品的评价报告必须按照规定的格式递交(SANCO/10597/2003-rev.9附件Ⅶ)。
4.4 评审成员国及认定流程 如果接收等同性认定申请的成员国(MS)不同意对该申请进行评价,则由比对产品的成员国(RMS)负责对该项申请进行审核。RMS需在接受该申请60天内对该项申请给出等同性认定报告,并将该报告告知欧盟委员会(COM)、其他成员国和申请者,后者需在30天之内对上述报告提出异议并反馈到RMS。如果对报告结论没有异议,RMS也需将相关结论告知其他成员国和欧盟委员会,同时后者负责告知欧盟食物链和动物健康常设委员会(SCFCAH)。如果MS和RMS之间未能对上述结论达成一致意见,也需要告知申请者、其他成员国和欧盟委员会,并说明原因。成员国之间应尽量就认定结论在45天之内达成一致意见。
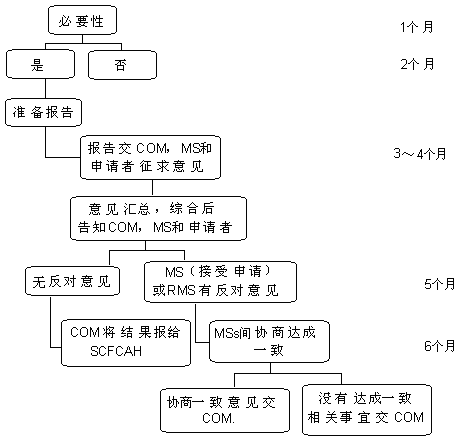
图1 等同性认定流程


|






















